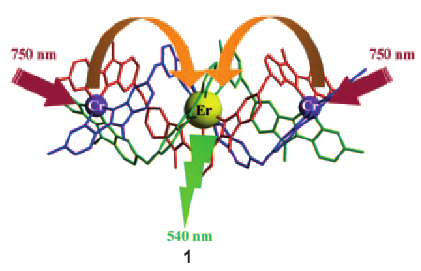-
Controlling Lanthanide Exchange in Triple-Stranded Helicates: A Way to Optimize Molecular Light-Upconversion
C. Piguet, D. Zare, Y. Suffren, H. Nozary and A. Hauser
Angewandte Chemie International Edition, 56 (46) (2017), p14612-14617


DOI:10.1002/anie.201709156 | unige:98963 | Abstract | Article HTML | Article PDF | Supporting Info
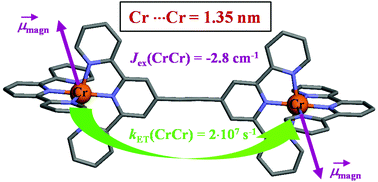
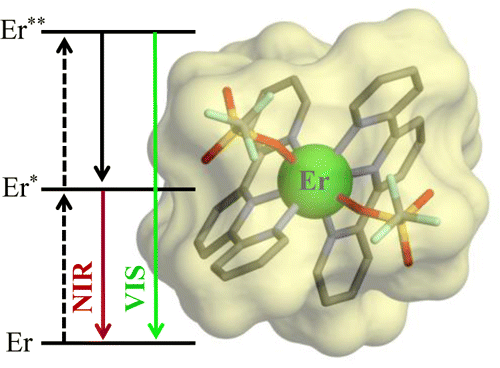
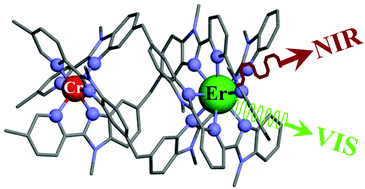
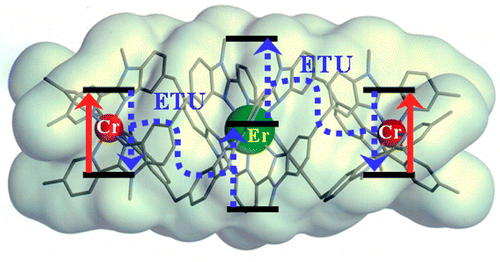
The kinetic lability of hexadentate gallium-based tripods is sufficient to ensure thermodynamic self-assembly of luminescent heterodimetallic [GaLn(L3)3]6+ helicates on the hour time scale, where Ln is a trivalent 4f-block cation. The inertness is however large enough for preserving the triple-helical structure when [GaLn(L3)3]6+ is exposed to lanthanide exchange. The connection of a second gallium-based tripod further slows down the exchange processes to such an extent that spectroscopically active [CrErCr(L4)3]9+ can be diluted into closed-shell [GaYGa(L4)3]9+ matrices without metal scrambling. This feature is exploited for pushing molecular-based energy transfer upconversion (ETU) at room temperature.